Lesedauer: 46 Minuten

Lesedauer: 46 Minuten

Entzündungsprozesse sind integraler Bestandteil der angeborenen Immunantwort und entscheidend für Gewebereparatur, Pathogenelimination und zelluläre Homöostase. Bei dysregulierter Aktivität können sie jedoch chronisch progrediente Schäden induzieren und gelten heute als zentrale pathogenetische Treiber zahlreicher Zivilisationskrankheiten – von kardiovaskulären Erkrankungen bis hin zu neurodegenerativen und autoimmunologischen Störungen. Genetische Polymorphismen in proinflammatorischen Signalwegen modulieren die individuelle Entzündungsantwort teils erheblich. Insbesondere Varianten in Genen wie IL6, TNF, CRP oder IL1B sind mit einer gesteigerten Zytokinproduktion und verlängerten inflammatorischen Aktivität assoziiert. Dieser Beitrag beleuchtet die molekulargenetischen Grundlagen einer erhöhten Entzündungsneigung, deren klinische Relevanz und die sich daraus ableitenden Implikationen für eine individualisierte Prävention und Therapie im Rahmen chronisch-inflammatorischer Erkrankungen.
Entzündungen gehören zu den grundlegendsten und zugleich komplexesten biologischen Reaktionsmechanismen des menschlichen Körpers. Sie stellen einen hochregulierten, evolutionär konservierten Prozess dar, mit dem der Organismus auf eine Vielzahl exogener und endogener Reize reagiert, darunter Infektionen, physikalische Verletzungen, toxische Substanzen, metabolischer Stress oder immunologische Dysregulation. Die primäre physiologische Aufgabe einer Entzündung besteht darin, eine potenzielle Schädigung zu begrenzen, eingedrungene Pathogene zu eliminieren, abgestorbene Zellen zu beseitigen und die Geweberegeneration einzuleiten. Eine korrekt ablaufende Entzündungsreaktion ist daher nicht nur wünschenswert, sondern essenziell für das Überleben.
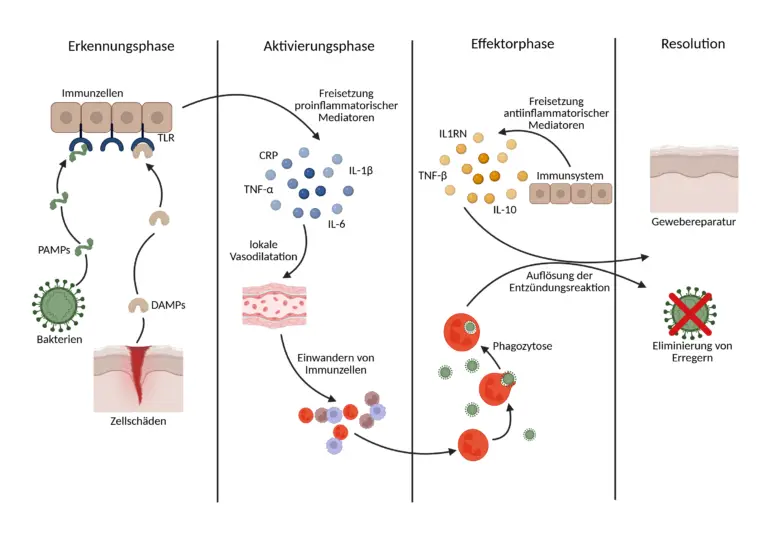
Die klassische Entzündungsantwort lässt sich in vier Phasen gliedern: Erkennung, Aktivierung, Effektorphase und Resolution. In der Erkennungsphase detektieren spezialisierte Immunzellen sogenannte Gefahrensignale, entweder pathogenassoziierte molekulare Muster (PAMPs) wie Lipopolysaccharide bakteriellen Ursprungs oder körpereigene, durch Zellschäden freigesetzte DAMPs (damage-associated molecular patterns). Diese Signale werden über Rezeptoren wie Toll-like Receptors (TLRs) erkannt, was zur Aktivierung intrazellulärer Signalwege führt. In der folgenden Aktivierungsphase kommt es zur raschen Freisetzung proinflammatorischer Mediatoren, darunter Zytokine wie TNF-α, IL-1β und IL-6, Chemokine, Prostaglandine und Leukotriene. Diese Moleküle orchestrieren eine lokale Vasodilatation, erhöhen die Permeabilität der Blutgefäße und ermöglichen das gezielte Einwandern von Immunzellen, insbesondere neutrophiler Granulozyten und Monozyten, in das betroffene Gewebe. In der Effektorphase eliminieren diese Zellen die auslösenden Schadfaktoren durch Phagozytose, enzymatische Degradation, oxidative Mechanismen und komplementvermittelte Prozesse. Gleichzeitig beginnt das Immunsystem, über antiinflammatorische Mediatoren (z. B. IL-10, TGF-β) und pro-resolutive Lipidmediatoren (z. B. Resolvine, Protectine) eine aktive Regulation und Auflösung der Entzündungsreaktion einzuleiten. Dieser Prozess ist essenziell für die Gewebshomöostase und die Vermeidung chronischer Schäden. Eine vollständige Resolution führt zur Gewebereparatur und zur Wiederherstellung der physiologischen Funktion.
Allerdings ist die Entzündungsreaktion ein zweischneidiges Schwert. Wenn die Rückregulation nicht adäquat funktioniert oder wenn persistierende Reize wie metabolischer Stress, Dysbiose, oxidativer Stress oder subklinische Infektionen vorliegen, kann die Entzündung chronisch werden. Diese niedriggradige, systemische Entzündungsaktivität, häufig als „silent inflammation“ oder „low-grade inflammation“ bezeichnet, verläuft meist symptomarm, zeigt jedoch eine anhaltende Aktivierung proinflammatorischer Signalwege und eine dauerhafte Präsenz inflammatorischer Biomarker im Gewebe und im Blut. Auf molekularer Ebene dominieren hier leicht erhöhte Spiegel von CRP, TNF-α, IL-6 sowie reaktive Sauerstoff- und Stickstoffspezies. Diese Form der chronischen Entzündung ist pathophysiologisch hochrelevant: Sie gilt heute als zentraler Risikofaktor für eine Vielzahl chronischer Erkrankungen, darunter kardiovaskuläre Erkrankungen, Typ-2-Diabetes, Adipositas, neurodegenerative Erkrankungen (z. B. Alzheimer, Parkinson), chronisch-entzündliche Darmerkrankungen und bestimmte Krebserkrankungen. Darüber hinaus wird sie mit dem Phänomen des „Inflammaging“ in Verbindung gebracht, einem durch chronische Entzündung beschleunigten Alterungsprozess, der zur funktionellen Verschlechterung von Organsystemen im höheren Lebensalter beiträgt. Ein entscheidender Aspekt, der über Ausmaß, Dauer und Schwere von Entzündungsreaktionen mitbestimmt, ist die genetische Prädisposition. Zahlreiche Studien belegen, dass Polymorphismen in zentralen Entzündungsgenen, insbesondere TNF-Alpha, IL6, IL6R, IL1RN und CRP, einen messbaren Einfluss auf die individuelle Entzündungsneigung haben. Diese Gene kodieren für Schlüsselmoleküle im Netzwerk der Immunregulation: TNF-Alpha und IL-6 sind primäre proinflammatorische Zytokine, IL6R beeinflusst deren Signaltransduktion, IL1RN wirkt als natürlicher Antagonist des Interleukin-1-Rezeptors, und CRP dient als systemischer Marker sowie potenzieller Verstärker entzündlicher Prozesse. Liegen diese Gene in bestimmten Varianten vor, etwa mit erhöhter Promotoraktivität oder veränderter Rezeptoraffinität, kann dies zu einer verstärkten oder verlängerten Entzündungsantwort führen. Diese genetischen Faktoren beeinflussen nicht nur das Entzündungsausmaß bei akuten Infektionen, sondern auch die Schwelle, ab der der Körper mit Entzündungen auf Alltagsreize wie Stress, Umweltbelastungen oder Ernährung reagiert.
Insbesondere bei Personen mit genetisch erhöhter Entzündungsbereitschaft können schon geringe, chronisch wirkende Reize zu einem permanenten Entzündungszustand führen. Diese stille Aktivierung ist häufig bereits im mittleren Lebensalter messbar, lange bevor klinische Symptome auftreten und stellt einen wesentlichen Ansatzpunkt für die personalisierte Prävention dar. Denn während die genetische Veranlagung nicht veränderbar ist, lässt sich ihr Einfluss durch gezielte präventivmedizinische Maßnahmen erheblich modulieren.
Das TNF-Alpha-Gen (Tumornekrosefaktor-Alpha, TNF) kodiert für das gleichnamige proinflammatorische Zytokin TNF-α, das eine zentrale Rolle in der Regulation immunologischer und entzündlicher Prozesse spielt. Das Gen befindet sich auf Chromosom 6, genauer im Bereich p21.3 (kurzer Arm des Chromosoms), innerhalb des hochregulierten MHC-Komplexes (Major Histocompatibility Complex), und umfasst etwa 3.000 Basenpaare. TNF-α wird primär von aktivierten Makrophagen, aber auch von anderen Immun- und Endothelzellen produziert und wirkt über parakrine und autokrine Signale auf zahlreiche Zielzellen im Immunsystem, im Gefäßendothel und im Fettgewebe. Die Produktion erfolgt typischerweise als Antwort auf immunologische Gefahrensignale, etwa bakterielle Lipopolysaccharide (LPS) oder zelluläre Stressfaktoren. TNF-α liegt zunächst in einer membrangebundenen Vorstufe (mTNF-α) vor, die durch das Enzym TACE (TNF-α converting enzyme) proteolytisch gespalten und in eine lösliche, zirkulierende Form (sTNF-α) überführt werden kann. Beide Formen sind biologisch aktiv und entfalten ihre Wirkung über zwei spezifische Zellrezeptoren: TNFR1 und TNFR2.
Die Bindung von TNF-α an TNFR1 führt zur Trimerisierung des Rezeptors und zur Rekrutierung intrazellulärer Adapterproteine wie TRADD, RIPK1, TRAF2 und FADD. Abhängig vom zellulären Kontext aktiviert dieser Komplex unterschiedliche Signalwege, insbesondere die Transkriptionsfaktoren NF-κB und AP-1. Die Aktivierung von NF-κB verläuft über die Phosphorylierung des Inhibitors IκB, dessen nachfolgender proteasomaler Abbau die Translokation von NF-κB in den Zellkern ermöglicht. Dort induziert NF-κB die Transkription zahlreicher Zielgene, die für die Produktion von proinflammatorischen Zytokinen (z. B. IL-1, IL-6), Chemokinen, Adhäsionsmolekülen, Enzymen wie COX-2 sowie antiapoptotischen Proteinen verantwortlich sind. Parallel kann TNF-α über denselben Rezeptor auch den extrinsischen Apoptoseweg aktivieren, insbesondere über die Aktivierung von Caspase-8 durch den FADD-Komplex. Diese Balance zwischen entzündungsfördernder Genexpression und programmiertem Zelltod ist hochgradig kontextabhängig und stellt ein zentrales Regulationsprinzip dar. Im Gegensatz dazu aktiviert TNFR2 primär nicht-apoptotische Signalwege und ist stärker an regenerativen und proliferativen Prozessen beteiligt. Über TRAF2 vermittelt TNFR2 die Aktivierung von MAPK-Kinasen, insbesondere p38 und JNK, was zur Zellproliferation, Angiogenese und Gewebeadaptation beiträgt. Eine Rolle, die vor allem bei chronischen Entzündungen und in der Wundheilung bedeutsam ist.
TNF-α entfaltet seine Wirkung jedoch nicht nur auf zellulärer Ebene, sondern wirkt als zentraler Taktgeber im systemischen Zytokinnetzwerk. Es verstärkt die Synthese weiterer Mediatoren wie IL-1, IL-6, GM-CSF und IFN-γ, erhöht die Gefäßpermeabilität, stimuliert die Expression von MHC-Molekülen auf Antigen-präsentierenden Zellen und erleichtert die Einwanderung von Immunzellen in das betroffene Gewebe. Darüber hinaus beeinflusst TNF-α auch den Stoffwechsel: In Adipozyten hemmt es die Insulin-Signaltransduktion durch Serinphosphorylierung des Insulinrezeptor-Substrats IRS-1, was zur Insulinresistenz beiträgt. Gleichzeitig fördert es die Lipolyse, erhöht die Konzentration freier Fettsäuren im Blut und begünstigt die systemische Inflammationslage.
Die Wirkung von TNF-α ist damit funktionell nicht auf eine einzelne Aufgabe beschränkt, sondern hochdynamisch und vielschichtig. Als frühes Alarmprotein des Immunsystems leitet es sowohl akute als auch chronische Entzündungsprozesse ein, reguliert Zellüberleben und -tod, vernetzt humorale und zelluläre Immunantworten und trägt wesentlich zur Steuerung des Gewebeumbaus bei. Diese Effekte sind fein austariert und zeitlich limitiert. Eine übermäßige oder anhaltende TNF-α-Aktivität kann jedoch zu Gewebeschäden, chronischer Entzündung und systemischer Dysregulation führen. Die biochemische Präzision, mit der TNF-α-Synthese, Sekretion, Rezeptorbindung und Signaltransduktion gesteuert werden, ist daher essenziell für die Aufrechterhaltung der Immunhomöostase und stellt einen zentralen Ansatzpunkt in der klinischen Immunologie und präventiven Medizin dar.
Damit Entzündungsreaktionen im Körper effizient gesteuert und nach ihrer Aufgabe auch wieder zuverlässig herunterreguliert werden können, braucht es eine funktionell ausbalancierte Aktivität des Zytokins Tumornekrosefaktor Alpha (TNF-α) und damit ein intaktes TNF-Alpha-Gen (TNF). Eine Schlüsselstelle für die Steuerung der Genexpression liegt im Promotorbereich des Gens beim Abschnitt rs1800629, an dem es zu einem Basenaustausch von Guanin (G) zu Adenin (A) kommen kann. Diese sogenannte Punktmutation beeinflusst die Transkriptionsrate des Gens und somit die Konzentration von TNF-α, einem der zentralen Entzündungsmediatoren im menschlichen Körper.
Im Rahmen der Genanalysen von NovoMedic kann durch die Bestimmung des TNF-Alpha-Genotyps (G/G, G/A und A/G oder A/A) die individuelle genetische Veranlagung zur Entzündungsregulation erfasst werden. Der G/G-Genotyp, der bei rund 83 % der mitteleuropäischen Bevölkerung vorkommt, stellt die ursprüngliche, funktionell ausgewogene Form des Gens dar. In diesem Fall wird TNF-α in physiologisch regulierter Menge produziert, das Immunsystem reagiert gezielt auf Bedrohungen und Entzündungsprozesse verlaufen in der Regel gut kontrolliert und selbstlimitierend. Die Immunantwort ist effizient, aber nicht übersteuert – eine zentrale Voraussetzung für langfristige Gewebegesundheit und ein balanciertes Entzündungsprofil. Der heterozygote Genotyp G/A oder A/G, der bei etwa 16 % der Bevölkerung auftritt, zeigt keine relevante Überproduktion von TNF-α, wird aber in wissenschaftlichen Studien als potenziell leicht reaktiver beschrieben, insbesondere unter chronischer Belastung oder in Verbindung mit weiteren proinflammatorischen Risikovarianten. In der klinischen Praxis zeigt sich jedoch meist eine normale Entzündungsregulation, insbesondere dann, wenn der Lebensstil entzündungsmodulierende Komponenten wie Bewegung, Schlaf, Mikronährstoffversorgung und antioxidative Ernährung umfasst. Die homozygote A/A-Variante, die nur bei rund 1 % der Bevölkerung nachweisbar ist, ist mit einer signifikant erhöhten TNF-Alpha-Expression assoziiert. In dieser genetischen Konstellation kann das Immunsystem auf Reize wie Stress, Infektionen oder Gewebeschäden überproportional stark reagieren. Die Folge ist eine aggressivere, länger andauernde Entzündungsaktivierung, die sich in einem erhöhten Basalniveau entzündlicher Marker und einer erhöhten Ausschüttung weiterer proinflammatorischer Zytokine wie IL-1 und IL-6 äußern kann. Personen mit dieser Genvariante zeigen eine verstärkte systemische Immunaktivität und sind besonders anfällig für chronische „stille“ Entzündungen (low-grade inflammation), vor allem bei gleichzeitig ungünstigem Lebensstil (z. B. Bewegungsmangel, viszerale Adipositas, hohe Zucker- oder Fettzufuhr, oxidativer Stress). In der Folge erhöht sich das Risiko für entzündungsgetriebene Erkrankungen wie Arteriosklerose, Insulinresistenz, nichtalkoholische Fettlebererkrankung, rheumatoide Beschwerden oder beschleunigte Alterungsprozesse (Inflammaging). Eine frühzeitige genetische Identifikation dieser Konstellation ermöglicht eine gezielte Prävention: etwa durch eine entzündungsarme Ernährung, mikronährstoffgestützte Immunmodulation (z. B. Omega-3-Fettsäuren, Vitamin D, Curcumin), regelmäßige Bewegung und Maßnahmen zur Stressreduktion.Das IL6-Gen (Interleukin-6, IL6) kodiert für das gleichnamige Zytokin Interleukin-6 (IL-6), das eine zentrale Rolle in der Schnittstelle zwischen angeborener und adaptiver Immunantwort spielt und an der Regulation zahlreicher physiologischer sowie pathophysiologischer Prozesse beteiligt ist. Das Gen befindet sich auf Chromosom 7 im Bereich p15.3–p21 (kurzer Arm des Chromosoms), umfasst etwa 5.000 Basenpaare und wird in verschiedenen Zelltypen exprimiert, darunter Makrophagen, T-Zellen, Endothelzellen, Fibroblasten, Adipozyten und Hepatozyten. Die Synthese von IL-6 wird insbesondere durch Entzündungsreize wie bakterielle Endotoxine (z. B. Lipopolysaccharide), virale RNA, gewebsständige Stresssignale sowie andere proinflammatorische Zytokine wie TNF-α und IL-1β induziert. IL-6 ist ein multifunktionelles, pleiotropes Zytokin, das sowohl proinflammatorische als auch antiinflammatorische Effekte entfalten kann – abhängig vom zellulären Kontext, der Dauer der Stimulation und der Art der Signalweiterleitung. Nach seiner Synthese wird IL-6 in den Extrazellularraum sezerniert und bindet dort an seinen spezifischen Rezeptorkomplex, der aus der IL-6-Rezeptor-Alpha-Kette (IL-6R) und dem Signaltransduktionsprotein gp130 besteht. Dabei wird zwischen zwei Signalwegen unterschieden: dem klassischen Signaling und dem trans-Signaling.
Beim klassischen Signaling bindet IL-6 an membranständige IL-6R-Moleküle, die hauptsächlich auf Hepatozyten, neutrophilen Granulozyten und einigen Leukozyten vorkommen. Nach Ligandenbindung bildet sich ein hexamerer Komplex mit gp130, der die intrazellulären Januskinasen (JAKs) aktiviert. Diese wiederum phosphorylieren den Transkriptionsfaktor STAT3, der in den Zellkern transloziert und dort die Expression zahlreicher Zielgene steuert. Darunter Akute-Phase-Proteine (z. B. CRP, Fibrinogen, Serumamyloid A), proinflammatorische Mediatoren sowie Gene der Zellproliferation, Angiogenese und Apoptoseregulation. Das trans-Signaling dagegen erfolgt über die Interaktion von IL-6 mit einem löslichen IL-6-Rezeptor (sIL-6R), der entweder durch proteolytische Spaltung oder alternatives Spleißen entsteht. Der entstehende IL-6/sIL-6R-Komplex kann dann auch Zellen erreichen, die keinen membranständigen IL-6R tragen, da gp130 auf nahezu allen Zelltypen exprimiert ist. Auf diese Weise entfaltet IL-6 über das trans-Signaling eine deutlich breitere und oft proinflammatorisch geprägte Wirkung, etwa auf Endothelzellen, glatte Muskelzellen oder Fibroblasten. Dieser Mechanismus spielt eine wichtige Rolle bei chronischen Entzündungen, Autoimmunprozessen und der Tumorprogression.
Besonders relevant ist IL-6 auch in seiner akut-phasischen Wirkung auf die Leber, wo es die Synthese von CRP, Fibrinogen, Haptoglobin und anderen Akute-Phase-Proteinen stimuliert. Gleichzeitig hemmt IL-6 die Expression von Albumin und Transferrin, ein Mechanismus, der dem Organismus hilft, Ressourcen kurzfristig auf die Immunabwehr zu konzentrieren. Im Bereich des adaptiven Immunsystems unterstützt IL-6 die Differenzierung von B-Zellen zu Antikörper-produzierenden Plasmazellen und fördert die Polarisierung von CD4+-T-Zellen zu Th17-Zellen, was bei Autoimmunerkrankungen eine pathogene Rolle spielen kann. Darüber hinaus beeinflusst IL-6 auch den metabolischen Stoffwechsel. In Adipozyten und Hepatozyten wirkt es insulinantagonistisch, fördert die Lipolyse, erhöht die Freisetzung freier Fettsäuren und trägt zur systemischen Insulinresistenz bei. IL-6 ist zudem an der zentralnervösen Regulation des Appetits, der Energiehomöostase und des Schlaf-Wach-Rhythmus beteiligt.
Die Wirkung von IL-6 ist damit funktionell nicht auf eine einzelne biologische Domäne beschränkt, sondern betrifft eine Vielzahl von Organsystemen und Regulationskreisen. Als zentrales Vermittlermolekül im Zytokinnetzwerk wirkt IL-6 sowohl lokal im Entzündungsherd als auch systemisch etwa über die Leber, das Fettgewebe oder das zentrale Nervensystem. Bei akuter, kontrollierter Aktivierung trägt IL-6 wesentlich zur Infektabwehr und Gewebereparatur bei. Eine dauerhafte Überaktivierung oder Dysregulation von IL-6 kann jedoch zur Entwicklung chronisch-inflammatorischer Erkrankungen führen. Darunter rheumatoide Arthritis, entzündliche Darmerkrankungen, Atherosklerose, Typ-2-Diabetes, Adipositas, chronische Fatigue und neurodegenerative Prozesse.
Die biochemische Feinabstimmung der IL-6-Produktion, -Sekretion, -Rezeptorbindung und -Signalweiterleitung ist daher entscheidend für die Aufrechterhaltung der Immun- und Stoffwechselhomöostase. Aufgrund dieser breiten und regulativen Effekte stellt IL-6 nicht nur einen zentralen diagnostischen Marker im Rahmen der Akut-Phase-Reaktion dar, sondern auch einen wichtigen therapeutischen Zielpunkt in der personalisierten Entzündungsmedizin.Damit Entzündungsprozesse im Körper präzise gesteuert, effektiv begrenzt und nach überstandener Immunantwort zuverlässig herunterreguliert werden können, braucht es eine fein abgestimmte Aktivität des Zytokins Interleukin-6 (IL-6) und damit ein funktionell reguliertes IL6-Gen. Eine Schlüsselrolle spielt hierbei der genetische Abschnitt rs1800795, an dem es zu einem Basenaustausch von Guanin (G) durch Cytosin (C) kommen kann. Diese sogenannte Punktmutation liegt im Promotorbereich des Gens und beeinflusst die Transkriptionsrate, also die Aktivität, mit der das IL6-Gen abgelesen wird. In der Folge kann sich die Menge an zirkulierendem IL-6 im Blut signifikant verändern mit Auswirkungen auf die Entzündungsregulation und das systemische Immunmilieu.
Im Rahmen der Genanalysen von NovoMedic kann durch die Bestimmung des IL6-Genotyps die individuelle genetische Veranlagung zur Entzündungsaktivität erfasst werden. Der G/G-Genotyp, der bei etwa 77 % der Bevölkerung vorkommt, entspricht der ursprünglichen, funktionell „normalen“ Form des Gens. In diesem Fall ist die IL-6-Produktion kontrolliert und entspricht der physiologischen Bedarfslage, etwa bei akuten Infekten oder Gewebeschäden. Das Immunsystem reagiert gezielt, lokal begrenzt und selbstlimitierend, ohne überschießende systemische Inflammationsreaktionen auszulösen. Entzündungsprozesse klingen in der Regel rasch wieder ab, und es entsteht kein dauerhaft erhöhter Entzündungszustand. Beim heterozygoten Genotyp G/C oder C/G, der bei rund 19 % der Bevölkerung auftritt, ist eines der beiden Allele verändert. Diese Variante ist mit einer erhöhten IL-6-Expression assoziiert, insbesondere bei Stress, Infektion oder metabolischer Belastung. In der Folge kann die Entzündungsreaktion intensiver oder länger andauernd verlaufen als nötig. Betroffene Personen neigen tendenziell zu einer verzögerten Rückregulation der Immunantwort, was langfristig zu einem chronisch leicht erhöhten IL-6-Spiegel im Blut führen kann. Diese stille Entzündungsaktivität (low-grade inflammation) wird heute mit der Entstehung zahlreicher chronischer Erkrankungen in Verbindung gebracht. Die homozygote C/C-Variante, die nur bei etwa 4 % der Bevölkerung vorliegt, ist mit einer besonders starken IL-6-Aktivität verknüpft. Studien zeigen, dass Personen mit diesem Genotyp bereits in Ruhe einen deutlich erhöhten Grundspiegel an IL-6 aufweisen können, unabhängig von akuten Erkrankungen. Die entzündliche Grundaktivität im Körper ist chronisch erhöht, was zu einer dauerhaften Reizung des Immunsystems führt. Diese Personen haben ein erhöhtes Risiko für chronisch-entzündliche Erkrankungen, insbesondere wenn zusätzliche Belastungsfaktoren wie Schlafmangel, oxidativer Stress, Übergewicht oder Bewegungsmangel hinzukommen. Bereits alltägliche Reize können zu übersteigerten Immunantworten führen, was langfristig zu einer systemischen Entgleisung der Immunhomöostase führen kann.
Durch die Bestimmung des IL6-Genotyps lässt sich daher präzise einschätzen, wie reaktiv das individuelle Entzündungssystem ist und wie gezielt man präventiv gegensteuern kann. Vor allem bei erhöhtem IL-6-Risiko sind entzündungsmodulierende Maßnahmen wie eine antientzündliche Ernährung, gezielte Mikronährstoffunterstützung (z. B. Omega-3-Fettsäuren, Vitamin D, Curcumin), ausreichend Bewegung und Stressreduktion besonders wichtig, um die Entzündungsregulation langfristig zu stabilisieren und das Risiko für chronische Erkrankungen zu senken.
Das IL1RN-Gen (Interleukin-1-Rezeptorantagonist, IL1RN) kodiert für das gleichnamige regulatorische Protein Interleukin-1-Rezeptorantagonist (IL-1Ra), das eine zentrale Rolle in der fein abgestimmten Kontrolle von Entzündungsreaktionen spielt. Das Gen befindet sich auf Chromosom 2, genauer im Bereich q14.2 (langer Arm des Chromosoms), in unmittelbarer Nähe zu anderen Genen der IL-1-Familie, darunter IL1A und IL1B. Es umfasst etwa 16.000 Basenpaare und wird in zahlreichen Geweben exprimiert, vor allem jedoch in Monozyten, Makrophagen, Endothelzellen, Hepatozyten und verschiedenen Epithelzelltypen. Die Synthese von IL-1Ra erfolgt meist als Reaktion auf entzündliche Reize, darunter TNF-α, IL-1β und Lipopolysaccharide (LPS) und dient dazu, überschießende oder potenziell schädliche Immunantworten gezielt einzudämmen.
IL-1Ra ist ein natürlicher Gegenspieler der proinflammatorischen Zytokine IL-1α und IL-1β, ohne selbst proinflammatorisch zu wirken. Es bindet kompetitiv an den Interleukin-1-Rezeptor Typ 1 (IL-1R1), ohne dabei eine intrazelluläre Signaltransduktion auszulösen. Auf diese Weise blockiert IL-1Ra die Aktivierung typischer Entzündungswege durch IL-1 und wirkt so als molekularer „Dämpfer“ in der Immunantwort. Der Rezeptor IL-1R1 ist weit verbreitet und kommt auf zahlreichen Zelltypen des Immunsystems sowie auf Endothelzellen, Hepatozyten, Chondrozyten und glatten Muskelzellen vor. Durch die Bindung von IL-1Ra an IL-1R1 wird die durch IL-1 ausgelöste Signalkaskade, darunter die Aktivierung von NF-κB, AP-1 und MAPK, gezielt unterbunden.
In physiologischen Entzündungsprozessen ist IL-1Ra essenziell für die Begrenzung von Gewebeschäden, die durch eine übermäßige oder verlängerte IL-1-Aktivität entstehen könnten. Besonders in der Akutphase trägt es dazu bei, die Balance zwischen pro- und antiinflammatorischen Kräften aufrechtzuerhalten. Die biologische Bedeutung von IL-1Ra zeigt sich auch daran, dass eine vollständige genetische Inaktivierung, wie bei seltenen autosomal-rezessiven Mutationen, zu schweren autoinflammatorischen Syndromen führen kann.
Neben der Blockade von IL-1-induzierter Zytokinfreisetzung wirkt IL-1Ra auch modulierend auf die Produktion von Akute-Phase-Proteinen in der Leber, auf die Aktivierung von Th17-Zellen, die Bildung von Adhäsionsmolekülen auf Endothelzellen sowie auf die Leukozytenmigration. Die Wirkung von IL-1Ra entfaltet sich also nicht über eine aktive Signalverstärkung, sondern über eine kompetitive Hemmung eines zentralen proinflammatorischen Pfades. Diese passiv-regulierende Funktion ist jedoch von enormer physiologischer Relevanz, insbesondere in Geweben mit hoher Zytokinaktivität, bei Gewebeumbauprozessen, Infekten oder immunologischen Reaktionen auf körperfremdes Material (z. B. Implantate). IL-1Ra bildet hier einen entscheidenden Schutzmechanismus gegen Entgleisungen des Immunsystems. Auch auf metabolischer Ebene spielt IL-1Ra eine wichtige Rolle: Durch die Blockade von IL-1β wirkt es insulinprotektiv, stabilisiert die Glukosetoleranz und reduziert die systemische Inflammationslage, insbesondere im Rahmen metabolischer Störungen wie Adipositas, Typ-2-Diabetes oder nichtalkoholischer Fettleber. Ein gestörtes Gleichgewicht zwischen IL-1β und IL-1Ra kann in solchen Kontexten zur Chronifizierung subklinischer Entzündungen beitragen.
Die Wirkung von IL-1Ra ist damit funktionell zentral für die Feinabstimmung der Immunantwort. Sie verhindert, dass aus einer notwendigen akuten Entzündung ein chronisch destruktiver Prozess wird. Die biochemische Kontrolle über IL1RN-Expression, IL-1Ra-Freisetzung, Rezeptorbindung und Verweildauer im Gewebe ist daher ein wesentlicher Baustein für die Aufrechterhaltung der Immunhomöostase. Dysregulationen dieses Systems sind heute mit einer Vielzahl chronisch-entzündlicher und autoimmuner Erkrankungen assoziiert, von rheumatoider Arthritis über chronisch-entzündliche Darmerkrankungen bis hin zu kardiovaskulären und metabolischen Krankheitsbildern. Die gezielte Analyse und therapeutische Beeinflussung der IL-1/IL-1Ra-Achse stellt daher einen zunehmend relevanten Ansatzpunkt in der personalisierten Immuntherapie und Präventionsmedizin dar.
Damit entzündliche Prozesse im Körper nicht außer Kontrolle geraten, sondern gezielt begrenzt und effektiv aufgelöst werden können, braucht es ein gut funktionierendes Gegengewicht zu proinflammatorischen Botenstoffen wie Interleukin-1β (IL-1β). Diese regulierende Rolle übernimmt das Protein Interleukin-1-Rezeptorantagonist (IL-1Ra), das durch das IL1RN-Gen kodiert wird. Entscheidend für die Wirksamkeit dieses Mechanismus ist unter anderem der genetische Abschnitt rs419598, bei dem es zu einem Basenaustausch von Cytosin (C) gegen Thymin (T) kommen kann. Diese Punktmutation beeinflusst die Menge an IL-1Ra, die im Körper produziert wird und damit die Fähigkeit, Entzündungen effektiv zu regulieren.
Im Rahmen der genetischen Analysen von NovoMedic kann durch die Bestimmung des IL1RN-Genotyps eingeschätzt werden, wie stark oder schwach diese natürliche Entzündungsbremse ausgeprägt ist. Der C/C-Genotyp, der bei etwa 5 % der Bevölkerung vorkommt, ist mit einer stabilen und ausgewogenen Regulation assoziiert. In dieser Konstellation ist ausreichend IL-1Ra vorhanden, um proinflammatorische Reize durch IL-1β wirkungsvoll abzuschwächen. Das Gleichgewicht zwischen entzündungsfördernden und entzündungshemmenden Prozessen ist in der Regel intakt. Personen mit dem heterozygoten T/C oder C/T-Genotyp, der bei rund 28 % der Bevölkerung nachweisbar ist, zeigen eine leicht reduzierte IL-1Ra-Produktion. Die Blockade von IL-1β-Signalen ist damit abgeschwächt, was zu einer höheren Reaktivität des Immunsystems führen kann, vor allem unter Belastung durch Stress, chronische Infektionen oder eine proinflammatorische Ernährung. Bei diesen Personen besteht ein erhöhtes Risiko für eine verzögerte Rückregulation von Entzündungen. Subklinisch kann sich dies durch persistierende Aktivität von Zytokinen wie IL-6 oder durch leicht erhöhte CRP-Werte äußern, ohne dass zwingend akute Symptome vorliegen. Der T/T-Genotyp, der bei etwa 67 % der Bevölkerung vorkommt, ist mit der niedrigsten IL-1Ra-Expression verbunden. In dieser genetischen Konstellation fehlt es dem Körper an ausreichender Gegenspielerkapazität, um die entzündungsfördernde Wirkung von IL-1β effizient zu kontrollieren. Die Folge kann eine chronisch erhöhte Entzündungsneigung sein. Ein Zustand, der in der Medizin auch als silent inflammation oder low-grade inflammation bezeichnet wird. Personen mit diesem Genotyp zeigen häufiger eine systemische Grundaktivierung des Immunsystems, selbst ohne akute Reize. Dies kann langfristig zur Entstehung oder Verschlechterung entzündungsgetriebener Erkrankungen beitragen.
Die Bestimmung des IL1RN-Genotyps liefert somit wertvolle Hinweise auf die individuelle Entzündungsregulation und bietet eine fundierte Grundlage für gezielte Präventionsmaßnahmen. Insbesondere bei genetisch erhöhter Entzündungsneigung empfiehlt sich ein antiinflammatorischer Lebensstil: eine ausgewogene, entzündungsmodulierende Ernährung, die gezielte Versorgung mit Mikronährstoffen wie Vitamin D, Omega-3-Fettsäuren oder Curcumin, regelmäßige Bewegung, guter Schlaf und effektives Stressmanagement. Auf diese Weise lässt sich das genetische Risiko gezielt entschärfen – bevor daraus langfristige Gesundheitsprobleme entstehen.
Das CRP-Gen (C-reaktives Protein) kodiert für das gleichnamige Akute-Phase-Protein CRP, das eine zentrale Rolle in der systemischen Entzündungsantwort des menschlichen Körpers spielt. Das Gen ist auf Chromosom 1 an der Position q23.2 (langer Arm des Chromosoms) lokalisiert und umfasst etwa 2.000 Basenpaare. Das von CRP codierte Protein wird überwiegend in Hepatozyten der Leber synthetisiert, insbesondere als Reaktion auf proinflammatorische Zytokine, allen voran Interleukin-6 (IL-6), aber auch IL-1β und TNF-α. CRP wird nach seiner Synthese rasch in den Blutkreislauf abgegeben und spielt dort eine Schlüsselrolle in der angeborenen Immunabwehr und als systemischer Marker entzündlicher Prozesse. Biochemisch handelt es sich bei CRP um ein Pentraxin, das aus fünf identischen Untereinheiten besteht, die sich zu einer ringförmigen Struktur zusammenlagern. Seine wichtigste Funktion besteht in der Erkennung und Markierung (Opsonierung) von Pathogenen oder zellulären Abbauprodukten. CRP bindet bevorzugt an Phosphocholinreste, die bei Zellschäden oder auf der Oberfläche grampositiver Bakterien vorkommen. Diese Bindung aktiviert das Komplementsystem, wodurch die Eliminierung markierter Strukturen durch Phagozyten erleichtert wird. Darüber hinaus wirkt CRP immunmodulatorisch, indem es die Ausschüttung proinflammatorischer Zytokine beeinflusst, die Leukozytenmigration reguliert und oxidativ modifizierte Lipoproteine markiert. In Gefäßwänden kann CRP die Expression von Adhäsionsmolekülen auf Endothelzellen erhöhen, die Aufnahme von LDL in Makrophagen fördern und so zur Entstehung atherosklerotischer Plaques beitragen, ein möglicher Mechanismus, durch den chronisch erhöhte CRP-Werte kardiovaskuläre Risikoprozesse mit beeinflussen. Die Plasmakonzentration von CRP unterliegt einer hochdynamischen Regulation: In Ruhe liegt sie meist unter 1 mg/L, bei akuter Entzündung kann sie jedoch innerhalb weniger Stunden um das bis zu 1000-Fache ansteigen. Diese Eigenschaft macht CRP zu einem empfindlichen Frühmarker systemischer Entzündungen. Auch niedriggradige, chronische lassen sich durch leicht erhöhte CRP-Werte im Bereich von 1–3 mg/L erfassen. Die Regulation der CRP-Expression ist jedoch nicht nur durch Entzündungsreize und hormonelle Faktoren gesteuert, sondern auch genetisch determiniert. Polymorphismen im Promotorbereich des CRP-Gens können die basale und stimulusabhängige Expression messbar beeinflussen. Personen mit genetisch bedingt reduzierter CRP-Expression weisen trotz aktiver Entzündungsprozesse unter Umständen nur leicht oder gar nicht erhöhte CRP-Spiegel im Blut auf, ein Umstand, der in der klinischen Interpretation zu Fehleinschätzungen führen kann. Umgekehrt können bestimmte genetische Varianten mit einer gesteigerten CRP-Produktion einhergehen, was unabhängig von der tatsächlichen Entzündungsaktivität zu dauerhaft erhöhten CRP-Werten führen kann. Eine gestörte CRP-Regulation, sei es durch genetische Varianten, epigenetische Veränderungen oder chronisch subklinische Entzündungsreize, kann somit zu einer fehlgeleiteten Immunaktivität und einer langfristigen Belastung der Gefäß- und Gewebsintegrität führen. In diesem Kontext kommt dem CRP-Gen auch präventivmedizinisch eine wachsende Bedeutung zu: Die Kenntnis über die individuelle CRP-Regulation erlaubt eine präzisere Einschätzung des tatsächlichen Entzündungsstatus und der damit assoziierten Risiken.
Das C-reaktive Protein (CRP) ist eines der wichtigsten Akutphasenproteine des menschlichen Körpers. Während seine Produktion in erster Linie durch Entzündungsbotenstoffe wie Interleukin-6 gesteuert wird, spielt auch die genetische Ausstattung eine entscheidende Rolle dabei, wie stark CRP im Körper reguliert wird.
Ein besonders relevanter Abschnitt im CRP-Gen ist der Polymorphismus rs3093066. An dieser Stelle kann es zu einem Basenaustausch von Thymin (T) zu Guanin (G) kommen. Eine scheinbar kleine Veränderung, die jedoch große Auswirkungen auf die CRP-Expression haben kann. Je nachdem, welche Genvariante vorliegt, fällt die Entzündungsbereitschaft des Körpers ganz unterschiedlich aus. Menschen mit dem T/T-Genotyp, der bei lediglich etwa 2 % der Bevölkerung vorkommt, verfügen über eine ruhige Entzündungsgrundlage. Die CRP-Produktion reagiert gezielt auf immunologische Reize und bleibt ansonsten auf niedrigem Niveau. Diese Genvariante begünstigt ein stabiles, ausgewogenes Entzündungsprofil – ein Vorteil, insbesondere im Hinblick auf das Risiko chronischer Entzündungsfolgen. Anders verhält es sich bei Personen mit einem T/G oder G/T-Genotyp, den etwa 11 % der Bevölkerung tragen. Hier zeigt sich eine höhere Grundaktivität des CRP-Gens: Schon bei moderatem Stress oder suboptimaler Lebensweise können sich messbar erhöhte CRP-Werte im Blut zeigen, auch ohne akuten Infekt. Diese subtil erhöhte Entzündungsbereitschaft bleibt oft lange unbemerkt, wird aber mit der Zeit zum Belastungsfaktor für Gefäße, Stoffwechsel und Immunsystem. Besonders häufig ist die G/G-Variante, die bei rund 87 % der Menschen vorliegt. In diesem Fall ist die genetisch bedingte CRP-Aktivität am stärksten ausgeprägt. Der Körper zeigt eine Tendenz zur Überreaktion auf Entzündungsreize, selbst bei geringfügigen Auslösern. Die Folge ist nicht selten ein dauerhaft leicht erhöhtes Entzündungsniveau im Blut und steht langfristig mit zahlreichen chronischen Erkrankungen in Verbindung: etwa Herz-Kreislauf-Erkrankungen, metabolisches Syndrom, nichtalkoholische Fettleber oder neurodegenerative Prozesse.
Die genetische Analyse des CRP-Gens bietet daher wertvolle Einblicke in die individuelle Entzündungstendenz. Nicht als isolierter Marker, sondern im Zusammenspiel mit anderen biologischen, lebensstilbezogenen und umweltbedingten Faktoren. Wer genetisch zu einer verstärkten CRP-Produktion neigt, profitiert besonders von präventiven Maßnahmen, die das Immunsystem im Gleichgewicht halten: eine antientzündliche Ernährung, regelmäßige Bewegung, eine ausgewogene Mikronährstoffversorgung und bewusste Stressbewältigung. Denn gerade dort, wo die genetische Grundausstattung zur Reizüberreaktion neigt, kann eine gezielte Lebensstilsteuerung entscheidend zur langfristigen Gesundheit beitragen.
Biochemisch handelt es sich bei CRP um ein Pentraxin, das aus fünf identischen Untereinheiten besteht, die sich zu einer ringförmigen Struktur zusammenlagern. Seine wichtigste Funktion besteht in der Erkennung und Markierung (Opsonierung) von Pathogenen oder zellulären Abbauprodukten. CRP bindet bevorzugt an Phosphocholinreste, die bei Zellschäden oder auf der Oberfläche grampositiver Bakterien vorkommen. Diese Bindung aktiviert das Komplementsystem, wodurch die Eliminierung markierter Strukturen durch Phagozyten erleichtert wird.
Darüber hinaus wirkt CRP immunmodulatorisch, indem es die Ausschüttung proinflammatorischer Zytokine beeinflusst, die Leukozytenmigration reguliert und oxidativ modifizierte Lipoproteine markiert. In Gefäßwänden kann CRP die Expression von Adhäsionsmolekülen auf Endothelzellen erhöhen, die Aufnahme von LDL in Makrophagen fördern und so zur Entstehung atherosklerotischer Plaques beitragen, ein möglicher Mechanismus, durch den chronisch erhöhte CRP-Werte kardiovaskuläre Risikoprozesse mit beeinflussen. Die Plasmakonzentration von CRP unterliegt einer hochdynamischen Regulation: In Ruhe liegt sie meist unter 1 mg/L, bei akuter Entzündung kann sie jedoch innerhalb weniger Stunden um das bis zu 1000-Fache ansteigen. Diese Eigenschaft macht CRP zu einem empfindlichen Frühmarker systemischer Entzündungen. Auch niedriggradige, chronische lassen sich durch leicht erhöhte CRP-Werte im Bereich von 1–3 mg/L erfassen.
Die Regulation der CRP-Expression ist jedoch nicht nur durch Entzündungsreize und hormonelle Faktoren gesteuert, sondern auch genetisch determiniert. Polymorphismen im Promotorbereich des CRP-Gens können die basale und stimulusabhängige Expression messbar beeinflussen. Personen mit genetisch bedingt reduzierter CRP-Expression weisen trotz aktiver Entzündungsprozesse unter Umständen nur leicht oder gar nicht erhöhte CRP-Spiegel im Blut auf, ein Umstand, der in der klinischen Interpretation zu Fehleinschätzungen führen kann. Umgekehrt können bestimmte genetische Varianten mit einer gesteigerten CRP-Produktion einhergehen, was unabhängig von der tatsächlichen Entzündungsaktivität zu dauerhaft erhöhten CRP-Werten führen kann. Eine gestörte CRP-Regulation, sei es durch genetische Varianten, epigenetische Veränderungen oder chronisch subklinische Entzündungsreize, kann somit zu einer fehlgeleiteten Immunaktivität und einer langfristigen Belastung der Gefäß- und Gewebsintegrität führen. In diesem Kontext kommt dem CRP-Gen auch präventivmedizinisch eine wachsende Bedeutung zu: Die Kenntnis über die individuelle CRP-Regulation erlaubt eine präzisere Einschätzung des tatsächlichen Entzündungsstatus und der damit assoziierten Risiken.
Das IL6R-Gen (Interleukin-6-Rezeptor) kodiert für die α-Untereinheit des Interleukin-6-Rezeptors (IL-6R), der eine zentrale Rolle in der Vermittlung der biologischen Wirkung von Interleukin-6 (IL-6) spielt, einem Schlüsselzytokin in der Entzündungsregulation, Immunabwehr und Stoffwechselkontrolle. Das Gen ist auf Chromosom 1 in der Region q21 lokalisiert und umfasst etwa 300.000 Basenpaare. Das von IL6R exprimierte Protein wird sowohl membrangebunden als auch in löslicher Form produziert und ist damit funktionell äußerst vielseitig: Es vermittelt sowohl klassische zellgebundene IL-6-Signale als auch sogenannte trans-signaling-Prozesse, die systemische Entzündungsreaktionen fördern.
Das IL-6R-Protein besteht aus einer extrazellulären Bindungsdomäne für IL-6, einer Transmembranregion sowie einem kurzen intrazellulären Abschnitt. Im klassischen Signalweg bindet IL-6 zunächst an den membrangebundenen IL-6R, der vor allem auf Leukozyten und Hepatozyten exprimiert ist. Diese Bindung ermöglicht die Rekrutierung des Signaltransduktionsmoleküls gp130, das nach Dimerisierung intrazelluläre Signalkaskaden aktiviert. Auf diese Weise steuert IL-6R die Genexpression zahlreicher entzündungs- und stoffwechselrelevanter Zielgene. Neben der klassischen Signalübertragung existiert ein zweiter, klinisch besonders relevanter Wirkmechanismus: das IL-6-Trans-Signaling. Hierbei bindet IL-6 an eine lösliche Form des IL-6-Rezeptors (sIL-6R), die durch proteolytische Spaltung des membrangebundenen Rezeptors oder durch alternatives Spleißen entsteht. Der IL-6/sIL-6R-Komplex kann mit gp130 auf nahezu allen Zelltypen interagieren, auch auf solchen, die keinen eigenen IL-6R exprimieren. Das Trans-Signaling ist besonders bei chronischen, systemischen Entzündungen aktiv und wird mit der Pathogenese zahlreicher Erkrankungen in Verbindung gebracht, Atherosklerose, Insulinresistenz, Fettleber, Tumorprogression oder neuroinflammatorische Prozesse.
Das Verhältnis zwischen membranständigem und löslichem IL-6R ist entscheidend für das Gleichgewicht zwischen regenerativen, lokal begrenzten und systemisch proinflammatorischen IL-6-Effekten. Eine erhöhte Konzentration von sIL-6R im Blut verstärkt die Reichweite und Intensität von IL-6-induzierten Signalen und trägt maßgeblich zur Entstehung chronischer „low-grade“-Entzündungen bei. Diese stille Entzündungsbereitschaft bleibt oft lange unentdeckt, ist aber als Risikofaktor für zahlreiche nicht übertragbare Erkrankungen etabliert.
Störungen in der Funktion oder Regulation des IL6R-Gens durch genetische Polymorphismen, epigenetische Veränderungen oder einseitige Daueraktivierung entzündlicher Signalwege können das empfindliche Gleichgewicht zwischen Schutz- und Risikomechanismen verschieben. Eine übermäßige IL-6R-Aktivität führt zu einer verlängerten Aktivierung proinflammatorischer Gene, einer erhöhten Zytokinfreisetzung und einer systemischen Entgleisung der Immunantwort. Gleichzeitig beeinflusst IL6R auch die Leberproduktion von Akute-Phase-Proteinen wie CRP und Serumamyloid A sowie die Differenzierung von T-Zell-Subtypen und die hämatopoetische Balance.
Die präzise Funktion des IL6R-Proteins ist daher essenziell für die Integrität der Entzündungsregulation, des Immunsystems und des Stoffwechsels. Sie stellt einen hochrelevanten Angriffspunkt in der personalisierten Medizin dar, sowohl diagnostisch als auch therapeutisch. Eine gezielte Analyse des IL6R-Gens kann helfen, individuelle Entzündungsrisiken frühzeitig zu erkennen und Maßnahmen zur Modulation der IL-6-Signalwege einzuleiten.
Ein zentraler Mechanismus, der darüber entscheidet, wie unser Körper auf entzündliche Reize reagiert, ist die Signalweiterleitung über den Interleukin-6-Rezeptor (IL-6R). Dieser Rezeptor vermittelt die Wirkung des Entzündungsbotenstoffs Interleukin-6 (IL-6), der nicht nur bei akuten Infektionen aktiv ist, sondern auch an stillen, chronischen Entzündungen beteiligt sein kann. Gesteuert wird die Herstellung des IL-6-Rezeptors durch das IL6R-Gen, dessen Aktivität unter anderem durch den genetischen Abschnitt rs2228145 beeinflusst wird. An dieser Stelle kann es zu einem Austausch der Base Adenin (A) gegen Cytosin (C) kommen, eine Veränderung, die direkte Auswirkungen auf die Menge und Art des Rezeptors im Körper hat.
Bei der genetischen Analyse im Rahmen der NovoMedic Diagnostik wird untersucht, welcher Genotyp an dieser Stelle vorliegt. Rund 52 % der Bevölkerung tragen die Ausprägung A/A. Diese Variante geht mit einem physiologisch ausbalancierten Verhältnis zwischen membranständigem und löslichem IL-6-Rezeptor einher. Entzündungsreaktionen können so zielgerichtet ausgelöst und nach getaner Arbeit auch zuverlässig heruntergefahren werden. Die Immunantwort bleibt lokal begrenzt und gut kontrolliert, ein ideales Szenario für die Aufrechterhaltung der entzündlichen Homöostase. Anders verhält es sich bei Personen mit der A/C oder C/A-Variante, die etwa 37 % der Bevölkerung betrifft. In dieser Konstellation führt das veränderte Allel dazu, dass mehr löslicher IL-6-Rezeptor (sIL-6R) gebildet wird. Dieser zirkuliert frei im Blut und kann gemeinsam mit IL-6 eine systemische Entzündungsaktivierung über das sogenannte Trans-Signaling auslösen, auch in Zelltypen, die von Natur aus keinen eigenen IL-6-Rezeptor tragen. Die Folge: Das Immunsystem wird breiter und potenziell länger aktiviert, selbst bei eher milden Reizen. Dieser genetische Hintergrund begünstigt somit eine erhöhte Reaktionsbereitschaft auf entzündungsfördernde Einflüsse. Bei rund 11 % der Bevölkerung liegt die C/C-Variante vor, die mit der höchsten Konzentration löslicher IL-6-Rezeptoren im Blut einhergeht. In dieser Konstellation ist die Entzündungstätigkeit besonders aktiv: Das IL-6-Trans-Signaling ist stark ausgeprägt und kann langfristig zu einer dauerhaft erhöhten Entzündungsgrundlage führen. Diese stille Entzündung wird heute als Mitursache zahlreicher chronischer Erkrankungen diskutiert, darunter Arteriosklerose, Typ-2-Diabetes, nichtalkoholische Fettleber, aber auch neurodegenerative Erkrankungen oder Autoimmunprozesse.
Die Kenntnis des individuellen IL6R-Genotyps kann deshalb ein wertvoller Baustein in der personalisierten Prävention sein. Gerade Personen mit A/C, C/A- oder C/C-Variante profitieren von einem gezielten Lebensstilmanagement, das auf eine Beruhigung des Immunsystems abzielt.
Überaktive oder chronisch unterschwellig verlaufende Entzündungsreaktionen gelten heute als einer der zentralen Risikofaktoren für eine Vielzahl moderner Erkrankungen, darunter Herz-Kreislauf-Leiden, Typ-2-Diabetes, nichtalkoholische Fettleber, Autoimmunprozesse und neurodegenerative Veränderungen. Wie intensiv und wie langanhaltend der Körper auf Entzündungsreize reagiert, ist dabei nicht allein vom Lebensstil abhängig, sondern auch genetisch mitbedingt. Varianten in Genen wie IL6, TNF, IL6R, IL1RN oder CRP können die Produktion, Aktivität oder Rückregulation entzündlicher Signalstoffe beeinflussen. Menschen mit entsprechender Veranlagung zeigen häufiger eine überdurchschnittlich starke Immunantwort, selbst auf alltägliche Reize wie Stress, bestimmte Nahrungsbestandteile oder Umweltfaktoren. Die Folge: eine dauerhafte, stille Aktivierung des Immunsystems, die auf zellulärer Ebene Schaden anrichtet, lange bevor klinische Symptome entstehen.
Umso wertvoller sind gezielte Präventionsstrategien, die helfen, das entzündliche Gleichgewicht im Körper wiederherzustellen. Besonders empfehlenswert ist eine antientzündliche Ernährung, die reich an buntem Gemüse, Beeren, grünen Blattgemüsen, Hülsenfrüchten, Nüssen, hochwertigem Olivenöl und fettem Meeresfisch ist. Reduziert werden sollten entzündungsfördernde Komponenten wie Zucker, Weißmehl, Alkohol, trans-Fette sowie stark verarbeitete Lebensmittel. Eine ausreichende Versorgung mit entzündungsmodulierenden Mikronährstoffen, darunter Omega-3-Fettsäuren (EPA/DHA), Vitamin D, Magnesium, Zink, aktive B-Vitamine und Vitamin C kann die Immunbalance zusätzlich unterstützen. Ebenso hilfreich sind sekundäre Pflanzenstoffe wie Curcumin, Resveratrol, Polyphenole aus grünem Tee, OPC oder Quercetin, die gezielt in Entzündungsprozesse eingreifen.
Auch Bewegung wirkt entzündungsregulierend, insbesondere moderates Ausdauertraining, Intervalltraining oder Yoga, das zusätzlich das parasympathische Nervensystem stärkt. Ein weiterer wichtiger Hebel ist ausreichender Schlaf, da viele proinflammatorische Zytokine einem zirkadianen Rhythmus folgen. Regelmäßige Essenspausen oder Formen des Intervallfastens tragen darüber hinaus dazu bei, entzündliche Signalwege herunterzufahren und das Immunsystem zu regenerieren.
Wer seine genetische Entzündungsneigung kennt, kann gezielt und frühzeitig gegensteuern. Nicht durch restriktiven Verzicht, sondern durch bewusste, alltagstaugliche Entscheidungen. So lassen sich überschießende Immunreaktionen eindämmen, stille Entzündungen verhindern und die langfristige Gesundheit von Gefäßen, Organen und Stoffwechsel nachhaltig schützen.
DocCheck Flexikon. (n.d.). Entzündung. https://flexikon.doccheck.com/de/Entz%C3%BCndung
Helmholtz-Zentrum für Infektionsforschung. (n.d.). Was ist eine Entzündung? https://www.helmholtz-hzi.de/wissen/wissensportal/entzuendung/
Lecturio. (n.d.). Entzündung – Definition, Phasen, Symptome. https://www.lecturio.de/artikel/medizin/entzundung/
Pinto-Sietsma, S. J., et al. (2005). Genetic variants and inflammation: Atherosclerosis context. PLoS ONE, 6(9), e69201. https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0069201
SNPedia. (n.d.). rs1800629 (TNF gene). https://www.snpedia.com/index.php/Rs1800629
Wang, M., et al. (2023). Tumor necrosis factor gene polymorphism and cardiovascular inflammation. Microorganisms, 12(10), 1954. https://www.mdpi.com/2076-2607/12/10/1954
Zhou, B., et al. (2012). Interactions between CRP and TNF-α gene variants. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677
Zhou, B., et al. (2012). IL6, IL6R, TNF, CRP gene interactions in inflammation. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677
Fishman, D., et al. (2002). The effect of a novel polymorphism in the interleukin-6 gene on IL-6 transcription and plasma levels. Human Molecular Genetics, 11(3), 289–297. https://academic.oup.com/hmg/article/11/3/289/634575
Pinto-Sietsma, S. J., et al. (2005). Genetic variants and inflammation: Atherosclerosis context. PLoS ONE, 6(9), e69201. https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0069201
SNPedia. (n.d.). rs1800795 (IL6 gene). https://www.snpedia.com/index.php/Rs1800795
Zhou, B., et al. (2012). Interactions between CRP and IL-6 gene variants. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677
Zhou, B., et al. (2012). IL6, IL6R, TNF, CRP gene interactions in inflammation. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677
SNPedia. (n.d.). rs419598 (IL1RN gene). https://www.snpedia.com/index.php/Rs419598
Ligthart, S., et al. (2016). Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. American Journal of Human Genetics, 103(5), 691–706. https://www.sciencedirect.com/science/article/abs/pii/S1043466621001605
Pinto-Sietsma, S. J., et al. (2005). Genetic variants and inflammation: Atherosclerosis context. PLoS ONE, 6(9), e69201. https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0069201
SNPedia. (n.d.). rs3093066 (CRP gene). https://www.snpedia.com/index.php/Rs3093066
Zhou, B., et al. (2012). Interactions between CRP and IL6, TNF gene variants. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677
Zhou, B., et al. (2012). IL6, IL6R, TNF, CRP gene interactions in inflammation. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677
Ferreira, R. C., et al. (2013). Functional IL6R genetic variants and risk of inflammatory diseases. PLoS Genetics, 9(4), e1003444. https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1003444
SNPedia. (n.d.). rs2228145 (IL6R gene). https://www.snpedia.com/index.php/Rs2228145
Zhou, B., et al. (2012). CRP gene interaction with IL6R SNPs. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677
Zhou, B., et al. (2012). IL6, IL6R, TNF, CRP gene interactions in inflammation. Scientific Reports, 2, 267. https://www.nature.com/articles/srep32677