Lesedauer: 21 Minuten

Lesedauer: 21 Minuten
Triglyceride sind zentrale Energieträger im menschlichen Stoffwechsel und werden nach der Nahrungsaufnahme in Form von Lipoproteinen durch den Blutkreislauf transportiert. Ihr Abbau und ihre Beseitigung erfordern ein präzises Zusammenspiel mehrerer Enzyme und Transportproteine, um eine übermäßige Akkumulation im Plasma zu verhindern. Zwei genetisch regulierte Apolipoproteine – Apolipoprotein A5 (APOA5) und Apolipoprotein E (APOE) – nehmen dabei Schlüsselfunktionen ein: APOA5 moduliert die Lipolyse triglyceridreicher Partikel durch Förderung der LPL-Aktivität, während APOE für die Erkennung und hepatische Aufnahme der Abbauprodukte verantwortlich ist. Genetische Varianten in diesen Genen können die Effizienz dieses Stoffwechselprozesses erheblich beeinflussen und so zur Entstehung einer Hypertriglyzeridämie beitragen – einem unabhängigen Risikofaktor für kardiometabolische Erkrankungen. Der folgende Beitrag beleuchtet die physiologischen Grundlagen, genetischen Unterschiede und praxisrelevanten Implikationen für Prävention und Therapie.
Triglyceride sind die häufigste Form von Fett im menschlichen Körper und stellen eine zentrale Energiequelle dar. Chemisch gesehen bestehen sie aus einem Glycerinmolekül, das mit drei Fettsäuren verestert ist – daher der Name Triglycerid. Diese Struktur macht sie wasserunlöslich, weshalb sie im Blut nicht frei, sondern eingebettet in sogenannte Lipoproteine transportiert werden. Nach einer Mahlzeit gelangen die über die Nahrung aufgenommenen Triglyceride in Form von Chylomikronen aus dem Darm in den Blutkreislauf. Zusätzlich produziert die Leber bei einem Energieüberschuss sogenannte VLDL-Partikel (very low density lipoproteins), die körpereigene Triglyceride transportieren. Im Blut dienen Triglyceride vor allem der Bereitstellung von Energie: Sie werden mithilfe des Enzyms Lipoproteinlipase (LPL) aus den Lipoproteinen herausgelöst, in freie Fettsäuren gespalten und anschließend in Muskel- oder Fettzellen aufgenommen. Dort können sie entweder unmittelbar zur Energiegewinnung genutzt oder als langfristige Reserve gespeichert werden. Ein Gramm Fett liefert mit etwa neun Kilokalorien mehr als doppelt so viel Energie wie Kohlenhydrate oder Eiweiß, weshalb Triglyceride einen besonders effektiven Energiespeicher darstellen. Die Konzentration von Triglyceriden im Blut wird durch ein fein abgestimmtes Zusammenspiel aus Nahrungsaufnahme, endogener Produktion, enzymatischem Abbau und Gewebeaufnahme reguliert. Kommt es jedoch zu einem Ungleichgewicht, etwa durch übermäßige Kalorienzufuhr, Bewegungsmangel, genetische Faktoren oder hormonelle Störungen, kann der Triglyceridspiegel im Blut dauerhaft erhöht sein. Eine solche Hypertriglyzeridämie gilt als unabhängiger Risikofaktor für eine Reihe von Erkrankungen, darunter die nichtalkoholische Fettleber (NAFLD), das metabolische Syndrom, Typ-2-Diabetes und kardiovaskuläre Komplikationen. Deshalb ist die Regulation von Triglyceriden in unserem Körper von maßgebender Bedeutung.
Der systemische Abbau und die anschließende hepatische Beseitigung sind komplexe Vorgänge, bei denen neben Enzymen wie der Lipoproteinlipase (LPL) insbesondere regulatorische Apolipoproteine eine zentrale Rolle spielen. Zwei dieser Schlüsselmoleküle, Apolipoprotein A5 (APOA5) und Apolipoprotein E (APOE), wirken über distinkte, aber eng koordinierte Mechanismen, um den effizienten Abbau und die zügige Entfernung triglyceridreicher Lipoproteine aus dem Kreislauf sicherzustellen.
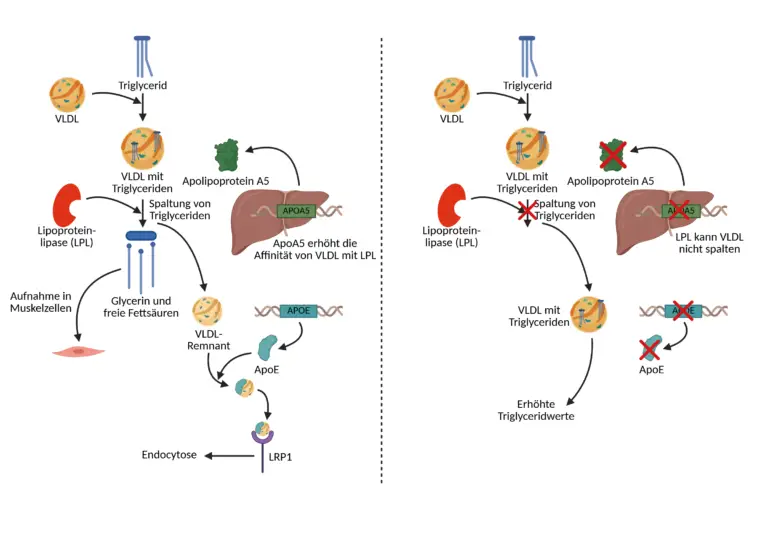
APOA5 ist ein leberspezifisch exprimiertes Apolipoprotein, das trotz seiner äußerst niedrigen Plasmakonzentration einen ausgeprägten Effekt auf die Höhe der zirkulierenden Triglyceride hat. Es übt seine Wirkung auf mehreren Ebenen aus: Zum einen erhöht APOA5 die Affinität triglyceridreicher Lipoproteine zur LPL, indem es deren Bindung an die Gefäßendothelien erleichtert und die Interaktion mit dem Enzym fördert. Dieser Schritt ist essenziell für die initiale Hydrolyse der Triglyceride in freie Fettsäuren und Glycerin, die anschließend in periphere Gewebe aufgenommen werden können. Zum anderen reduziert APOA5 die hepatische Sekretion von VLDL-Partikeln, indem es die Verpackung von Triglyceriden in ApoB-haltige Lipoproteine intrazellulär hemmt. Darüber hinaus verändert APOA5 die Oberflächenstruktur der Lipoproteine so, dass ihre Erkennung und Aufnahme durch hepatische Rezeptoren erleichtert wird. Diese Mehrfachwirkung positioniert APOA5 als zentralen Modulator des intravaskulären Triglyceridabbaus und der Lipoproteinfreisetzung.
APOE hingegen wirkt vorrangig in der Endphase des Triglyceridtransports. Als Ligand für spezifische hepatische Rezeptoren, insbesondere für das LDL-receptor-related protein 1 (LRP1) und weitere hepatozelluläre Aufnahmesysteme, vermittelt es die gezielte Aufnahme von Chylomikronen- und VLDL-Remnants in die Leber. Diese Partikel enthalten nach enzymatischer Lipolyse durch LPL nur noch geringe Mengen an Triglyceriden, müssen aber rasch aus dem Kreislauf entfernt werden, um eine Akkumulation zu verhindern. APOE fungiert hier als molekulares Erkennungssignal auf der Partikeloberfläche und interagiert mit zellulären Rezeptorkomplexen, um die Remnant-Aufnahme zu beschleunigen. Zusätzlich stabilisiert APOE die Lipoproteinstruktur und verbessert ihre Interaktion mit peripheren Enzymen und zellulären Aufnahmesystemen, wodurch die Effizienz des gesamten Triglyceridabbaus weiter optimiert wird.
Das Zusammenspiel von APOA5 und APOE ist funktionell hochkomplementär und entscheidend für eine physiologisch ausgewogene Triglyceridkonzentration im Blut. Während APOA5 den frühen Abbauschritt der Triglyceride durch LPL initiiert und die Bildung triglyceridreicher Lipoproteine begrenzt, übernimmt APOE die Verantwortung für die nachgelagerte Entfernung der verstoffwechselten Lipidreste. In gewisser Weise stellt APOA5 die „Startsequenz“ für den Triglyceridabbau dar, während APOE den „Abschluss“ organisiert. Beide agieren in unterschiedlichen Phasen desselben Stoffwechselweges, aber in enger funktioneller Abstimmung. Ihre koordinierte Wirkung gewährleistet, dass die postprandial entstehenden Lipoproteine rasch abgebaut, umgewandelt und aus dem Kreislauf entfernt werden können.
Eine Störung in einem dieser Regelmechanismen, etwa durch genetische Varianten, inflammatorische Prozesse oder ein Überangebot an Nahrungsfetten, kann zu einer verzögerten Triglycerid-Beseitigung führen. Die molekulare Aufklärung der Rolle von APOA5 und APOE bietet somit wertvolle Ansatzpunkte für eine personalisierte Prävention, Diagnostik und Therapie von Fettstoffwechselstörungen.
Das APOA5-Gen (Apolipoprotein A5) kodiert für das gleichnamige Apolipoprotein A5 (APOA5), das eine zentrale Rolle in der Regulation des Triglyceridstoffwechsels spielt. Das Gen ist auf Chromosom 11 an der Position q23 (langer Arm des Chromosoms) lokalisiert und umfasst etwa 30.000 Basenpaare. Das von APOA5 exprimierte Protein wird hauptsächlich in der Leber synthetisiert und in den Blutkreislauf abgegeben, wo es an der Oberfläche triglyceridreicher Lipoproteine wie Chylomikronen und VLDL (very low density lipoproteins) lokalisiert ist. APOA5 wirkt nicht strukturell, sondern regulierend auf die Kinetik des Triglyceridabbaus und die Verweildauer dieser Partikel im Blut.
Ein zentrales Enzym im intravaskulären Abbau von Triglyceriden ist die Lipoproteinlipase (LPL). Sie katalysiert die Hydrolyse von Triglyceriden innerhalb der Lipoproteine zu Glycerin und freien Fettsäuren, die dann in Muskel- oder Fettzellen aufgenommen werden können. APOA5 hat keinen direkten katalytischen Einfluss auf diese Spaltung, moduliert jedoch entscheidend die Effizienz der Lipolyse: Es erhöht die Affinität von Chylomikronen und VLDL-Partikeln zur LPL, indem es deren Bindung an endothelial gebundene Heparansulfat Proteoglykane stabilisiert und die Partikelstruktur so verändert, dass LPL optimal angreifen kann. In Anwesenheit von APOA5 steigt die Lipolysegeschwindigkeit signifikant, was zu einer schnelleren Entfernung von Triglyceriden aus dem Blut führt.
Darüber hinaus reguliert APOA5 auch die hepatische Sekretion von VLDL-Partikeln, indem es intrazellulär die Verpackung von Triglyceriden in ApoB-haltige Lipoproteine moduliert. Eine erhöhte APOA5-Expression ist mit einer verminderten VLDL-Sekretion und damit mit einer reduzierten Triglyceridlast im Blut assoziiert. Parallel dazu erleichtert APOA5 die hepatische Aufnahme von Remnants, also der Partikelreste nach erfolgter Lipolyse, wodurch die Triglycerid-Restmengen effizient aus dem Kreislauf entfernt werden. Die Wirkung von APOA5 ist dabei nicht statisch, sondern hochdynamisch und eng mit dem postprandialen Stoffwechsel verknüpft. Nach einer fettreichen Mahlzeit steigt der Bedarf an effizienter Triglycerid-Beseitigung erheblich. APOA5 ist unter diesen Bedingungen essenziell, um eine übermäßige Lipidakkumulation im Blut zu verhindern. Das Protein agiert als funktioneller Katalysator der Triglyceridregulation, indem es sowohl die enzymatische Hydrolyse als auch die Partikelbeseitigung beschleunigt und die Lipoproteinfreisetzung moduliert.
Eine gestörte Funktion des APOA5-Gens, beispielsweise durch genetische Polymorphismen oder epigenetische Veränderungen, kann die Aktivität dieses Regulationssystems erheblich beeinträchtigen. In der Folge kommt es zu einer verzögerten Lipolyse, einer verlängerten zirkulatorischen Verweildauer triglyceridreicher Partikel und einem erhöhten Triglyceridspiegel im Plasma. Die präzise Funktion von APOA5 ist daher entscheidend für die Integrität des Triglyceridstoffwechsels und stellt einen potenziellen therapeutischen und präventiven Angriffspunkt in der personalisierten Medizin dar.
Damit Triglyceride im Blut effizient abgebaut und reguliert werden können, braucht es ein funktionell aktives Apolipoprotein A5 und damit ein intaktes APOA5-Gen. Eine Schlüsselrolle spielt dabei der genetische Abschnitt rs662799, bei dem es zu einem Austausch der Base Adenin (A) durch Guanin (G) kommen kann. Diese Punktmutation beeinflusst die Genexpression und kann die Konzentration und Funktionalität von Apolipoprotein A5 im Blut deutlich verändern.
Im Rahmen der Genanalysen von NovoMedic kann durch die Bestimmung des APOA5-Genotyps (A/A, A/G oder G/A und G/G) die individuelle genetische Veranlagung zur Triglyceridregulation erfasst werden. Die Genvariante A/A, die bei etwa 71 % der Bevölkerung vorkommt, stellt die ursprüngliche, funktionell günstige Form des Gens dar. In diesem Fall ist die Expression von Apolipoprotein A5 ausreichend aktiv, sodass eine effektive Triglyceridregulation möglich ist. Die Triglyceride werden effizient durch die Lipoproteinlipase gespalten, und die VLDL-Sekretion bleibt auf physiologischem Niveau. Beim heterozygoten Genotyp A/G oder G/A, der bei rund 26 % der Bevölkerung auftritt, ist eines der beiden Allele verändert. Dies kann zu einer reduzierten Expression von APOA5 führen. In der Folge funktioniert die Triglycerid-Beseitigung weniger effizient, die Bindung triglyceridreicher Lipoproteine an die Lipoproteinlipase ist abgeschwächt und die Verweildauer von VLDL-Partikeln im Blut verlängert. Personen mit diesem Genotyp neigen tendenziell zu erhöhten Nüchtern-Triglyceridwerten, vor allem dann, wenn die Ernährung reich an einfachen Kohlenhydraten oder gesättigten Fetten ist. Die homozygote Risikovariante G/G, die nur bei etwa 3 % der Bevölkerung vorliegt, ist mit einer deutlich verringerten APOA5-Expression verbunden. Die Folge ist eine messbar gestörte Fähigkeit, Triglyceride aus dem Blut zu entfernen. Das Zusammenspiel mit der Lipoproteinlipase ist deutlich beeinträchtigt, wodurch triglyceridreiche Lipoproteine länger im Blut verbleiben. Betroffene Personen weisen häufig bereits bei normaler Ernährung eine erhöhte Triglyceridkonzentration im Plasma auf und haben ein erhöhtes Risiko für metabolische Folgeerkrankungen wie nichtalkoholische Fettleber, Insulinresistenz oder kardiovaskuläre Erkrankungen.
Das APOE-Gen (Apolipoprotein E) kodiert für das gleichnamige Protein Apolipoprotein E (APOE), das eine zentrale Rolle im Lipidstoffwechsel spielt, insbesondere bei der Regulation von Triglyceriden im Blutplasma. Das Gen ist auf Chromosom 19 an der Position q13.32 (langer Arm des Chromosoms) lokalisiert und umfasst rund 3.600 Basenpaare. Das von APOE codierte Protein wird vor allem in der Leber, aber auch im Gehirn und in Makrophagen synthetisiert. Es ist ein strukturmodifizierendes und rezeptorvermittelndes Apolipoprotein, das sich auf der Oberfläche verschiedener Lipoproteine wie Chylomikronen, VLDL und deren Abbauprodukten, sogenannten Remnants, befindet. Seine zentrale Funktion liegt in der Rezeptor-vermittelten Aufnahme triglyceridreicher Lipoproteinreste durch die Leber, wodurch es direkt in die Beseitigung von Triglyceriden eingebunden ist.
Nach der Nahrungsaufnahme zirkulieren Chylomikronen und VLDL-Partikel durch den Blutkreislauf und geben mithilfe der Lipoproteinlipase (LPL) ihre Triglyceridladung an periphere Gewebe ab. Zurück bleiben Triglycerid-reduzierte Partikel, sogenannte Remnants, die in ihrer Zusammensetzung und Struktur verändert sind. Diese Partikel können vom Organismus nur dann effizient entfernt werden, wenn sie von Rezeptoren in der Leber erkannt und aufgenommen werden und genau an dieser Stelle ist APOE unverzichtbar. Es fungiert als Ligand für verschiedene hepatische Lipoproteinrezeptoren, unter anderem den LDL-receptor-related protein 1 (LRP1) sowie hepatische Heparansulfat-Proteoglykane (HSPG). Durch diese Ligandenfunktion ermöglicht APOE die Erkennung und Endozytose der Remnants durch Hepatozyten. Auf diese Weise trägt APOE maßgeblich zur Reduktion der Triglyceridreste im Plasma bei. Während der Lipolyse- und Remnantbildung kann sich die Lipoproteinstruktur dynamisch verändern, denn es entstehen instabile, potenziell atherogene Partikel, deren sichere Beseitigung vom funktionellen Zustand des Apolipoprotein E abhängt. APOE stabilisiert die Oberfläche dieser Partikel und bereitet sie strukturell auf die Interaktion mit zellulären Rezeptoren vor. Zudem ermöglicht es durch seine amphipathische Helixstruktur die korrekte Orientierung der Lipoproteine an der Zellmembran, was die Bindungswahrscheinlichkeit und die Aufnahmerate zusätzlich erhöht.
Dieser Zyklus aus Triglyceridabgabe, Partikelumwandlung, APOE-vermittelter Erkennung und hepatischer Aufnahme ist hochdynamisch und läuft kontinuierlich ab, vor allem nach fettreichen Mahlzeiten. Die Regelmäßigkeit und Effizienz dieses Vorgangs hängt direkt von der funktionellen Integrität des APOE-Proteins ab. Ohne eine funktionierende APOE-Bindung würden Remnants unzureichend erkannt, länger im Blut verbleiben und zu einer erhöhten zirkulatorischen Triglyceridkonzentration führen. Die Folge wäre eine chronische Belastung des Lipidstoffwechsels und ein erhöhtes Risiko für Hypertriglyzeridämie, Fettleber, Insulinresistenz sowie kardiovaskuläre Erkrankungen.
Eine gestörte APOE-Funktion, etwa durch genetische Polymorphismen, die die Proteinstruktur oder Rezeptoraffinität verändern, führt zu einer verminderten Effizienz bei der Aufnahme von Remnants und somit zu einer Verlangsamung der Triglyceridclearance. Die daraus resultierende Akkumulation triglyceridreicher Partikel im Plasma kann langfristig zu einer Lipotoxizität führen, die sowohl vaskuläre als auch metabolische Komplikationen begünstigt.
APOE vermittelt die Erkennung triglyceridreicher Lipoproteine durch spezifische Rezeptoren in der Leber und unterstützt damit ihre zügige Aufnahme und Verstoffwechslung. Kommt es dabei zu Störungen, können sich Triglyceride im Blut anreichern, was das Risiko für verschiedene Stoffwechselerkrankungen erhöht.
Das Gen liegt in drei häufigen Varianten (Allelen) vor: ε2, ε3 und ε4. Diese unterscheiden sich durch minimale Veränderungen im genetischen Code, die dazu führen, dass an bestimmten Stellen im Protein unterschiedliche Aminosäuren eingebaut werden. Diese strukturellen Abweichungen beeinflussen, wie gut das Protein mit Rezeptoren interagieren kann. Die Kombination aus den beiden vererbten Allelen ergibt den individuellen APOE-Genotyp und dieser hat direkten Einfluss darauf, wie effizient der Triglyceridabbau im Körper abläuft.
Mit Hilfe der genetischen Analysen von NovoMedic kann genau bestimmt werden, welche APOE-Variante bei einer Person vorliegt. Das erlaubt Rückschlüsse auf die individuelle Neigung zu erhöhten oder normalen Triglyceridwerten. Der Genotyp ε2/ε2, der bei etwa 1 % der Bevölkerung vorkommt, geht häufig mit einer eingeschränkten Fähigkeit zur Aufnahme triglyceridreicher Lipoproteinreste in der Leber einher. Das bedeutet: Diese Partikel zirkulieren länger im Blut, und Triglyceride werden langsamer abgebaut. Betroffene zeigen daher eine genetische Tendenz zu erhöhten Triglyceridwerten, vor allem dann, wenn keine kompensierenden Lebensstilmaßnahmen gesetzt werden. Auch Personen mit dem Genotyp ε2/ε3 (etwa 6 % der Bevölkerung) verfügen über eine APOE-Variante mit eingeschränkter Rezeptorbindung. Obwohl das ε3-Allel einen gewissen Ausgleich schafft, zeigt sich in der Praxis häufig ebenfalls eine vererbte Neigung zu leicht erhöhten Triglyceriden. Der Genotyp ε3/ε3 ist mit rund zwei Drittel der Bevölkerung der häufigste und gilt in Bezug auf den Triglyceridstoffwechsel als neutral. Die ApoE-Struktur erlaubt eine effiziente Aufnahme von Remnants, sodass Triglyceride in der Regel gut reguliert werden. Eine Besonderheit stellt der Genotyp ε2/ε4 dar, der bei rund 2 % der Bevölkerung beobachtet wird. Hier treffen zwei unterschiedlich wirkende Varianten aufeinander: Das ε2-Allel mit schwächerer Rezeptorbindung und das ε4-Allel mit veränderter Konformation. In Kombination gleichen sich diese Eigenschaften in vielen Fällen aus, sodass die Fähigkeit zur Triglyceridregulation meist im Normbereich liegt, allerdings können Umwelteinflüsse diesen Ausgleich stören. Beim Genotyp ε3/ε4, der etwa ein Viertel der Bevölkerung betrifft, zeigt sich eine erhöhte genetische Veranlagung zu erhöhten Triglyceridwerten. Die ε4-Variante kann die Bindungseffizienz von ApoE herabsetzen, was zu einer verlangsamten Entfernung triglyceridreicher Lipoproteine aus dem Blut führt. Noch ausgeprägter ist dieser Effekt beim seltenen Genotyp ε4/ε4, den nur rund 1 % der Menschen tragen. Hier liegen zwei ε4-Allele vor, was zu einer deutlich eingeschränkten Funktion des ApoE-Proteins führen kann. Die Folge ist eine deutlich verlangsamte Triglycerid-Beseitigung, was sich in dauerhaft erhöhten Blutwerten niederschlagen kann.
Die genetische Analyse des APOE-Gens liefert somit wertvolle Hinweise für eine gezielte Prävention: Sie zeigt auf, ob eine genetische Neigung zu erhöhten Triglyceridwerten besteht und kann dabei helfen, passende Strategien in Ernährung, Bewegung und Mikronährstoffversorgung individuell abzustimmen.
Erhöhte Triglyceridwerte sind ein bedeutender Risikofaktor für Stoffwechselstörungen, Fettleber und Herz-Kreislauf-Erkrankungen. Genetische Varianten in den Genen APOA5 und APOE können beeinflussen, wie effizient der Körper Triglyceride abbaut und aus dem Blut entfernt. Bei entsprechender Veranlagung fällt es dem Organismus schwerer, nach fettreichen oder zuckerreichen Mahlzeiten überschüssige Blutfette rasch zu verarbeiten. Das macht gezielte Präventionsmaßnahmen besonders wertvoll.
Empfehlenswert ist vor allem eine pflanzenbasierte, ballaststoffreiche Ernährung mit viel Gemüse, Hülsenfrüchten, Nüssen, Beeren und hochwertigen Ölen. Zucker, Weißmehl, stark verarbeitete Lebensmittel sowie Alkohol sollten reduziert werden, da sie die körpereigene VLDL-Produktion erhöhen können. Mikronährstoffe wie Omega-3-Fettsäuren (EPA/DHA), L-Carnitin, Niacin, Magnesium, Zink, Vitamin E und aktive B-Vitamine können den Fettstoffwechsel zusätzlich stabilisieren. Auch Polyphenole aus grünem Tee, Curcumin oder OPC wirken entzündungshemmend und unterstützen die Gefäßgesundheit. Zentral ist außerdem ein aktiver Lebensstil: regelmäßige Bewegung, insbesondere Ausdauer- und Intervalltraining, verbessert die Fettverbrennung und senkt die VLDL-Freisetzung. Ergänzend helfen regelmäßige Essenspausen (z. B. Intervallfasten), um die Triglyceridsynthese zu drosseln und den Stoffwechsel zu entlasten.
Wer seine genetische Veranlagung kennt, kann gezielt gegensteuern, nicht durch Verzicht, sondern durch bewusste Entscheidungen. So lassen sich Triglyceridwerte effektiv stabilisieren und die langfristige Stoffwechselgesundheit fördern.
Afonso, M. S., & Alves, G. L. (2021). Genetic variants in lipid metabolism: Impact on disease risk and therapy. Frontiers in Neuroscience, 15, 630502. https://doi.org/10.3389/fnins.2021.630502
(https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2021.630502/full)
Calabresi, L., Pisciotta, L., & Tassi, V. (2018). Lipid metabolism and kidney disease: Insights from genetic studies. BMC Nephrology, 23, 291. https://doi.org/10.1186/s12882-022-02925-1
(https://bmcnephrol.biomedcentral.com/articles/10.1186/s12882-022-02925-1)
Clee, S. M., & Gould, R. M. (2012). Apolipoproteins: Structure, function, and clinical implications. The Journal of Lipid Research, 53(4), 599-613. https://doi.org/10.1194/jlr.R022531
(https://www.jlr.org/article/S0022-2275%2820%2935467-5/fulltext)
Lee, J. Y., & Lee, M. H. (2023). Advances in understanding apolipoprotein regulation. FEBS Letters, 597(5), 669-681. https://doi.org/10.1002/1873-3468.14803
(https://febs.onlinelibrary.wiley.com/doi/10.1002/1873-3468.14803)
Teramoto, T., & Kajinami, K. (2007). Role of Apolipoprotein in dyslipidemia. Yonsei Medical Journal, 48(4), 609-617. https://doi.org/10.3349/ymj.2007.48.4.609
(https://eymj.org/DOIx.php?id=10.3349%2Fymj.2007.48.4.609)
DocCheck Flexikon. (n.d.). Apolipoprotein A5. https://flexikon.doccheck.com/de/Apolipoprotein_A5
GeneCards. (n.d.). APOA5 gene. https://www.genecards.org/cgi-bin/carddisp.pl?gene=APOA5
Pennacchio, L. A., Olivier, M., Hubacek, J. A., et al. (2013). An Apolipoprotein A5 gene variant is associated with plasma triglyceride levels. PLoS ONE, 8(11), e56216. https://doi.org/10.1371/journal.pone.0056216
(https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0056216)
Pennacchio, L. A., Olivier, M., Hubacek, J. A., et al. (2016). ApoA5 variants and their impact on lipid metabolism. Scientific Reports, 6, 36830. https://doi.org/10.1038/srep36830
(https://www.nature.com/articles/srep36830)
PharmGKB. (n.d.). Clinical Annotation for rs662799. https://www.pharmgkb.org/clinicalAnnotation/1183492035
SNPedia. (n.d.). Rs662799. https://www.snpedia.com/index.php/Rs662799
Vega, A., Moreno, F., et al. (2018). APOA5 genetic variants and cardiovascular disease risk. The Journal of Clinical Lipidology, 12(6), 1352-1362. https://doi.org/10.1016/j.jacl.2018.07.002
(https://www.sciencedirect.com/science/article/abs/pii/S0031302518305245)
Wang, J., et al. (2020). The APOA5 rs662799 polymorphism and lipid levels: A meta-analysis. PLoS ONE, 7(3), e29385. https://doi.org/10.1371/journal.pone.0029385
(https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0029385)
Weinberg, R., et al. (2023). Apolipoprotein A5 polymorphisms in metabolic syndrome. Nutrients, 14(12), 2427. https://doi.org/10.3390/nu14122427
(https://www.mdpi.com/2072-6643/14/12/2427)
Zong, G., et al. (2021). APOA5 gene and triglyceride levels in East Asian populations. Journal of Lipid Research, 65(6), 100800. https://doi.org/10.1016/j.jlr.2023.100800
(https://www.jlr.org/article/S0022-2275(24)00083-X/fulltext)
NCBI Gene. (n.d.). APOE gene: Location, function and variants. https://www.ncbi.nlm.nih.gov/gene/116519
Lu, Y., & Chen, J. (2014). Apolipoprotein E alleles and risk of Alzheimer’s disease: A meta-analysis. Scientific Reports, 4, 5825. https://doi.org/10.1038/srep05825
(https://www.nature.com/articles/srep18184)
Lee, J. H., et al. (2013). APOE genotype and lipid metabolism. Atherosclerosis, 230(2), 184-190. https://doi.org/10.1016/j.atherosclerosis.2013.05.014
(https://pubmed.ncbi.nlm.nih.gov/24943010/)
PMC (2021). Apolipoprotein E polymorphisms and neurodegeneration. PMC, 8378982. https://doi.org/10.3390/ijms22168669
(https://pmc.ncbi.nlm.nih.gov/articles/PMC8378982/)